
HTML
-
油气是干酪根热解生烃的产物,其热解机理一直是受广泛关注的重要基础研究领域。根据化学反应动力学的时温补偿原理,可以在实验室通过提高模拟温度,缩短反应时间的方法来模拟有机质在地质条件下低温漫长的烃类生成过程。通过这种实验模拟的方法,一方面,为研究干酪根生烃动力学机制、计算反应速率、计算生烃量和确定生烃演化历史等提供了可靠的实验与理论依据[1];另一方面,对比了温度[2⁃3]、升温速率[4⁃6]、压力[2,7]、水介质[8⁃9]和催化剂[10⁃13]等因素对热解生烃模拟实验的影响。然而,传统的干酪根生烃模拟实验不仅面临反应条件复杂、难以确定分解温度等诸多制约因素,而且由于缺乏有效的检测工具来检测不断变化的自由基和中间体,导致在实验室条件下研究干酪根的详细反应过程和机理相当困难[14]。分子模拟是一种利用计算机技术在原子水平上模拟分子结构和行为,进而分析分子体系的各种物理化学性质的方法[15],其逐渐成为理解在分子结构中发生复杂反应基本原理的有力手段。因此,将分子模拟应用于干酪根热解,可以在原子和分子水平上研究干酪根的性质和生烃过程,并进一步提高对干酪根热解机理的认识。
本文在查阅大量文献的基础上,介绍了干酪根的分类和热解特征、干酪根分子结构模型研究现状及分子模拟研究方法,并将利用分子模拟技术对干酪根进行热解模拟取得的一些认识进行了重点详述,提出了存在的不足和未来的发展方向,以期对我国油气勘探提供有益帮助。
-
沉积物中有机质的主要贡献者是浮游植物、细菌、高等植物和浮游动物[16],它们基本上都是由糖类、脂类、蛋白质及木质素所组成[16⁃17]。这些由生物合成的天然有机质一旦进入沉积物中就成为沉积有机质,但最初的生物聚合体大部分在微生物的参与下很快被分解为各种单体(氨基酸、单糖等),未被消耗的单体在微生物的进一步作用下经活泼基团重新聚合成为较高相对分子质量的低聚物—腐殖质,腐殖质进一步缩聚、脱水、去官能团成为聚合度更高、稳定性更高的地质聚合物——干酪根[16⁃17]。前人对干酪根的定义是分散在沉积岩中不溶于有机溶剂、非氧化性酸的分散有机质的总称[16,18],主要由C、H、O和少量S、N等元素组成[19]。
根据H/C和O/C原子比,干酪根主要分为三类。I型干酪根的H/C原子比一般大于1.5,而O/C原子比一般小于0.1,芳香族和杂原子结构含量低,但富含脂肪链,其抽提物、油或热解产物富含长链的正构烷烃,碳数可大于C40[18]。II型干酪根的H/C原子比一般介于1.0~1.3,O/C原子比在0.15左右,相比于I型干酪根,含有更多的脂肪环以及芳香族结构,抽提物或热解产物中的正构烷烃分布几乎全部小于C25,即使在低成熟度下也是如此[18]。III型干酪根的H/C原子比一般小于0.8,O/C原子比可达0.3,其结构中含有大量的脂肪环和芳香族化合物[18,20]。
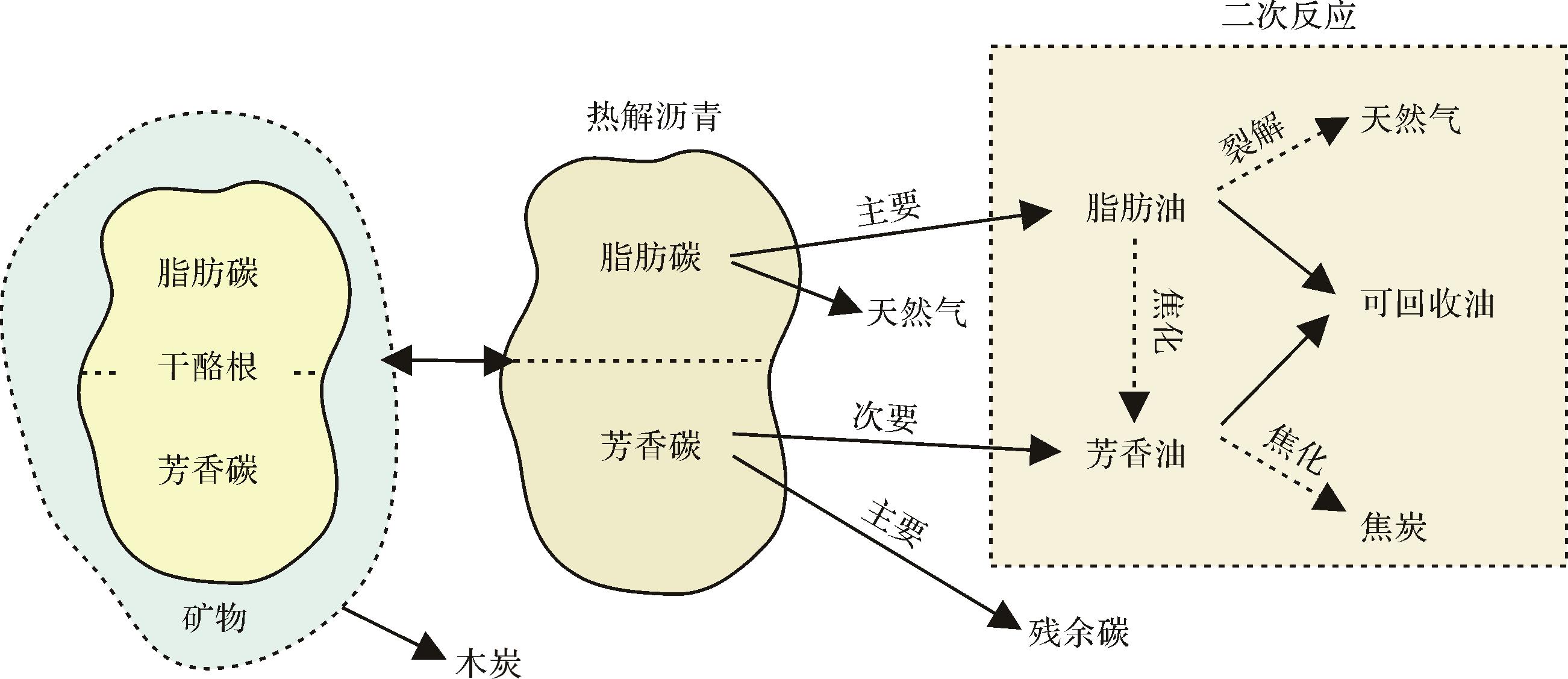
干酪根生成石油和天然气的潜力取决于干酪根的化学结构[21⁃22]。Miknis et al.[23⁃25]通过固体13C NMR发现,总脂肪碳与费舍尔方法测定的产油率之间存在很强的相关性,而芳香族碳主要与残余碳的产率有关,对产油率的贡献很小。这表明干酪根的油气生成潜力取决于其中的脂肪族碳,而不是芳香族碳[20]。在此基础上,Lai et al.[20]提出了干酪根热解过程中碳转化的机理模型(图1)。在最初的热解反应中,干酪根通过丢失少量碎片并保留基本的三维结构,从而形成流体和可溶的中间产物即热解沥青。在二次反应中,热解沥青中的脂肪族部分主要形成脂肪油,脂肪油可进一步裂解形成天然气或者焦化形成芳香油。而热解沥青中的芳香族部分大部分结焦形成残余碳,只有少量形成芳香油,芳香油可进一步焦化形成焦炭。最终的可回收油由干酪根热解形成的脂肪油和芳香油组成。

Figure 1. Mechanism model of carbon transformation during kerogen pyrolysis (modified from reference [20])
Behar et al.[26⁃27]对湖相Ⅰ型干酪根(绿河页岩)、海相Ⅱ型干酪根(托尔阶页岩)和Ⅲ型褐煤进行了无水封闭体系热模拟实验,其热解产物按化学类别划分为含N、S和O的重质化合物(NSOs)以及总的碳氢化合物(HCs)。根据这两类热模拟产物的变化提出了干酪根的热解机制:干酪根首先裂解成含有少量HC的NSOs,然后二氯甲烷NSOs和正戊烷NSOs先后连续裂解产生大量HC(即干酪根→NSOs→HCs),这证实了1969年Tissot[28]提出的石油形成模型。另外,Lai et al.[20]和Behar et al.[26⁃27]提出的机制都强调了沥青或NSOs的中间热解产物,事实上这两种干酪根连续热解生烃的机制是相同的,因为沥青富含NSOs,而石油富含HCs[29]。
Behar et al.[26]研究表明,对于干酪根的经典连续反应生油气的模型,不同类型的干酪根其生油气过程存在差异。随着非烃气体的释放,干酪根固体残余物的量开始减少,II型干酪根转化率(TR)高于40%时,固体残余物产率达到一个相对恒定的低值,然后增加,而III型干酪根在TR为70%~80%时达到最低值。对于II型干酪根和III型褐煤来说,一旦开始裂解就会产生二氯甲烷NSOs。II型干酪根产生的二氯甲烷NSOs在25%~70%的TR范围内达到稳定的最高值,而III型褐煤在TR为50%左右时产生的二氯甲烷NSOs达到最高值,之后均随着TR的增大而下降。与二氯甲烷NSOs相比,II型干酪根的正戊烷NSOs产率在更高的TR范围(45%~90%)达到稳定的最高值,III型褐煤则在较低的TR范围(30%~50%)正戊烷NSOs产率达到最高值。
除上述干酪根类型外,由于沉积环境和生物贡献的不同,还分布着另一种干酪根:湖相Ⅱ型干酪根[29]。Ma et al.[29]和Song et al.[30]对含有湖湘II型干酪根的鄂尔多斯盆地长7段和松辽盆地青山口组页岩进行了半封闭体系热模拟实验,发现在干酪根分解过程中,具有同时生成沥青和石油、同时生成NSOs和HCs的特征,大部分石油来自干酪根的热解,只有少部分来自沥青的热解。这与Lai et al.[20]和Behar et al.[26⁃27]描述的干酪根→沥青→石油或干酪根→NSOs→碳氢化合物的连续反应机制相矛盾,但证实了Burnham et al.[31]提出的“替代途径”机制,即油气可以从干酪根中与NSOs直接平行形成,并且这两类产物的形成由彼此独立的断键反应控制。
-
对干酪根进行分子模拟的研究主要是基于所建立的二维或三维干酪根分子结构模型。干酪根是多种大分子有机化合物组成的混合物,具有非常复杂的分子结构和广泛的分子质量范围,没有固定的化学式和分子结构,所以不可能构建完全一致的干酪根分子模型[1]。因此,“平均分子结构”的概念已被用来描述干酪根分子的最大可能结构,以克服构建干酪根结构模型的困难,目前这一概念已经广泛用于煤、油页岩有机质、石油重质油及各种沥青的化学结构研究[1,19]。
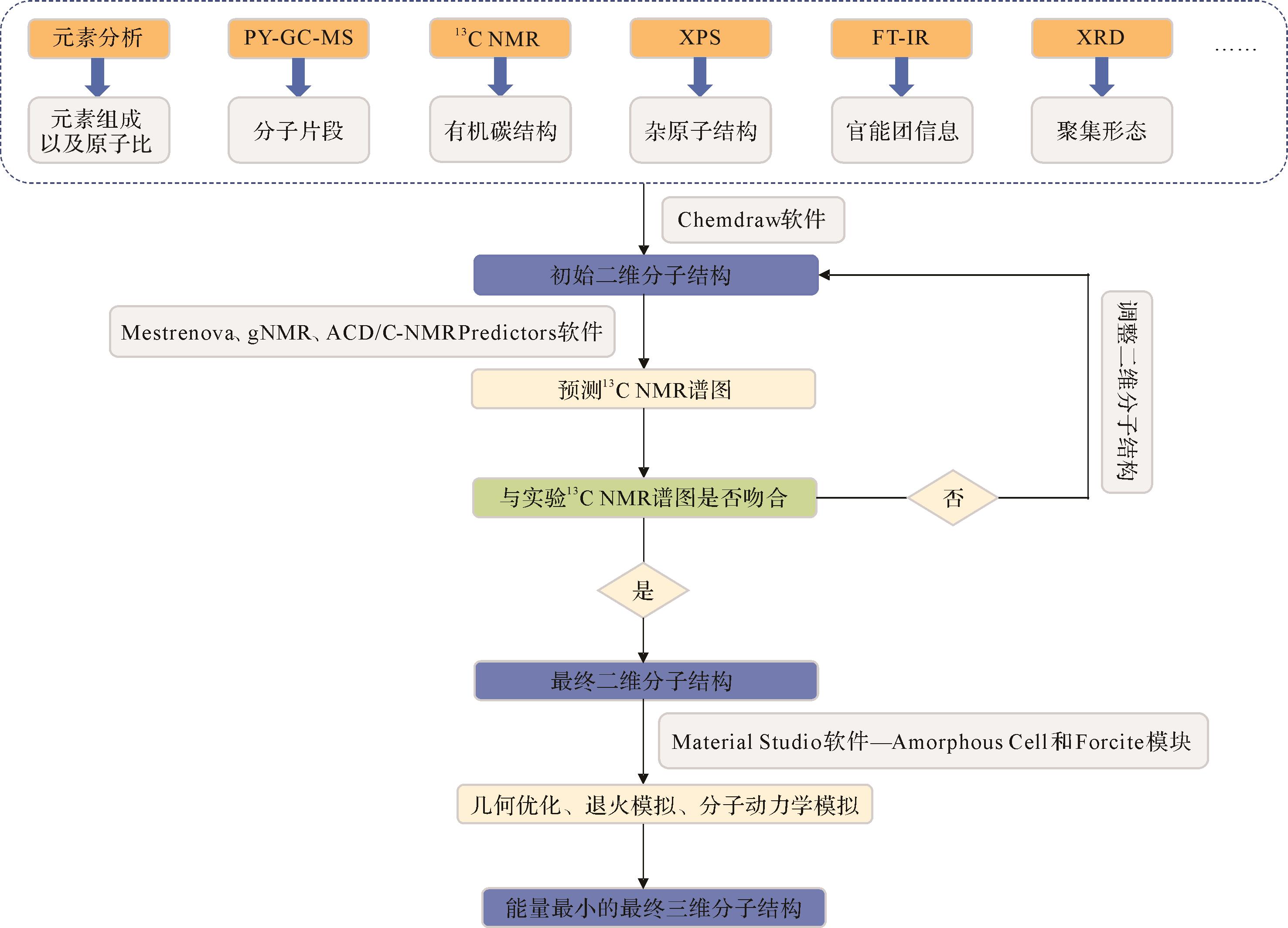
目前用于构建干酪根分子结构模型的方法通常有三种[32]。图2显示了基于实验分析来构建干酪根分子结构的第一种方法:首先,通过元素分析(EA)、热解气相色谱质谱(PY-GC-MS)、固体13C核磁共振(13C NMR)、X射线光电子能谱(XPS)、傅里叶变换红外光谱(FT-IR)和X射线衍射(XRD)等实验方法[19]对干酪根的化学结构进行表征,获得的元素、官能团信息和结构参数用于构建干酪根的初始二维分子结构;其次,通过调整脂肪族/芳香族区域碳骨架和杂原子官能团的位置使得模拟13C NMR谱图和实验谱图基本吻合,从而验证二维分子结构模型的合理性。最后,通过几何优化、退火模拟、分子动力学模拟对二维模型进行充分调整和优化,将能量最低的稳定结构作为最终的干酪根三维分子结构模型。第二种是重建法,即最初构成干酪根的原子随机分布,然后使用分子模拟方法重新定位,形成分子结构模型[32],例如Obliger et al.[33]和Bousige et al.[34]分别使用淬火分子动力学方法和分子动力学—混合逆向蒙特卡罗(MD-HRMC)方法来重建干酪根模型。第三种是通过木质素和纤维素等陆生植物成分的降解和重组来模拟干酪根的地质形成过程,从而建立干酪根模型[35⁃36]。相比于其他两种方法,第一种方法能够更加准确地反映干酪根的元素和官能团组成,这使得在应用于如干酪根热成熟和热解等此类以化学过程和产物为重点的模拟时能够提供比较可靠的数据[32]。此外,这也是建立干酪根和煤模型最常用的方法。

Figure 2. Process for constructing the kerogen structure model using experimental analysis
前人针对各个地区不同类型干酪根建立的结构模型分子式(表1)。著名的油页岩干酪根分子模型之一是Siskin et al.[37]建立的绿河油页岩I型干酪根模型,其分子式为C645H1017N19O17S4,它是通过13C NMR、29Si NMR和热解技术所构建的二维结构模型,其结构参数可以很好地与实验测定值吻合,并且已经进一步构建了三维结构模型[38]。由于干酪根类型、成熟度不同,使得干酪根的分子结构存在差别,因此前人基于各种实验分析,建立了不同类型和成熟度的干酪根分子模型。Behar et al.[48]基于EA、FT-IR、13C NMR等测试分析技术建立了美国绿河页岩(I型干酪根)、法国下托阿尔统页岩(II型干酪根)和喀麦隆杜阿拉盆地上白垩统沉积岩(III型干酪根)三种不同成熟度和类型的干酪根模型,其中在成岩阶段初期的干酪根模型分子质量均在25 000 Da左右(因为Behar et al. [48]建立的干酪根只有分子结构图片,没有分子式,且文献较早,分子结构不直观,因此在表1中并没有列出其分子式)。Ungerer et al.[39]根据Kelemen et al.[49]的实验分析数据构建了6种不同类型和成熟度的油页岩三维干酪根模型,并对其体积和热力学性质进行了定量预测,发现预测的干酪根密度和热容与其热成熟度和有机质类型具有很好的一致性。这六种干酪根模型也被后来的研究者用于进一步研究页岩气在干酪根中的吸附[50]以及成熟度[51]、含水率[51⁃52]和盐度[52]对吸附的影响。
干酪根 分子结构 干酪根类型 作者 绿河油页岩干酪根 C645H1017N19O17S4 I型 Siskin et al.[37],Orendt et al.[38] 未成熟绿河油页岩干酪根 C251H385O13N7S3 I-A型 Ungerer et al.[39] 未成熟迪韦奈干酪根 C252H294O24N6S3 II-A型 油窗顶部迪韦奈干酪根 C234H263O14N5S2 II-B型 油窗中—末期迪韦奈干酪根 C242H219O13N5S2 II-C型 过成熟迪韦奈干酪根 C175H102O9N4S2 II-D型 未成熟亚洲地区干酪根 C233H204O27N4 III-A型 桦甸油页岩干酪根 C243H407N3O25S2 I型 茹鑫[19] C235H365O25N3S3 I型 Guan et al.[40] C387H683O30N5S4 I型 Tong et al.[41⁃42] 甘肃窑街干酪根 C249H257N5O12S2 II型 Wang et al.[43] 松辽盆地干酪根 C612H846N10O40S7 I型 Wang et al.[44] 鄂尔多斯盆地干酪根 C853H756N8O115S6 III型 长7段油页岩干酪根 C300H297O18N13S10 II型 Wu et al.[45] 龙口油页岩干酪根 C341H481O40N5S6 II型 Zhang et al.[46] 抚顺油页岩干酪根 C228H350O11N6S3 II型 Zhao et al.[47] Table 1. Molecular structure model of kerogen
随着国内对油页岩研究的兴起,人们为进一步探索各地油页岩干酪根的差别以及生烃特性,针对国内各地的油页岩建立了专属的干酪根模型。茹鑫[19]利用13C NMR、PY-GC-MS、FT-IR等测试手段对吉林桦甸油页岩干酪根的分子结构进行了统计学分析,在建立的合理桦甸油页岩干酪根二维分子结构模型(C243H407N3O25S2)基础上进一步构建了三维模型,并通过计算键的Mayer键级和原子的Mulliken电荷数分析了模型的反应活性位点,推测出干酪根分子结构在实际升温过程中的分解步骤。与茹鑫[19]不同的是,Guan et al.[40]和Tong et al.[41⁃42]分别构建了12个和6个桦甸油页岩干酪根二维分子结构的构型异构体,其分子式分别为C235H365O25N3S3和C387H683O30N5S4,并最终得到一个能量最小并且模型与实验的13C NMR核磁共振谱吻合良好的三维模型。Wang et al.[43]采用分子模拟方法和多种测试技术相结合的方法,从构建的12个不同网格的甘肃窑街干酪根二维模型的异构体中确定了合理的三维模型(C249H257N5O12S2)。在他们建立模型的过程中,都提到了“网格”的概念,即由脂族链相互连接或与芳香族簇连接而构成的闭合环状结构[32]。干酪根的总能量取决于网格的数量[19,53],选择不同的网格可以明显增加或降低干酪根分子模型的能量。因此在建模过程通过设计合理的网格数,可以使得干酪根的总能量达到最低[40]。
就以往所建立的干酪根模型而言,除了C、H、O原子外,均引入了杂原子S和N,这些原子通过各种共价键、分子内非成键原子或分子间的范德华力和氢键连接在一起,分子质量最高达到25 000 Da[48],最终形成以核、桥键、官能团、侧链及被包裹组分为主的基本结构组分[17]。
对于干酪根生烃机理的分子模拟而言,对构建的干酪根分子模型的精度可能要求更高一些,以S原子为例,Lewan[54]研究发现C-S键的键能低于C-C键,富含S的II型干酪根生烃早于一般的干酪根,因此在构建干酪根分子模型的时候,必须尽可能地考虑更多元素的影响,官能团的构成、各个基团的构成及连接方式等。
大多数学者针对干酪根热解的分子模拟研究仅基于一个干酪根模型,例如Wang et al.[44]研究了温度(2 000 ℃、2 500 ℃、3 000 ℃)对单个松辽盆地I型干酪根(C612H846N10O40S7)和鄂尔多斯III型干酪根(C853H756N8O115S6)热解产物的影响。目前,越来越多的研究者倾向于对多个干酪根分子组成的大规模体系进行热解研究。Qian et al.[14]构建了包含10个Siskin绿河油页岩干酪根模型[37]的大规模体系,其中共包含17 160个原子,以此来研究油页岩干酪根非等温热解过程中的化学机理和产物。也有许多研究者基于Ungerer et al.[39]提出的模型,建立了不同成熟度不同类型的干酪根大规模体系,其中包含不同的干酪根分子数,以此来研究干酪根成熟过程中的交联途径[55]、原位转化过程中的热解行为[56]和干酪根热解机制与动力学[57]。Wu et al.[45]基于PY-GC-MS、13C NMR和XPS建立了分别包含3个和5个长7段油页岩II型干酪根分子(C300H297O18N13S10)的大规模体系,以此进一步研究干酪根的产物分布。Zhang et al.[46,58⁃60]基于建立的龙口油页岩II型干酪根模型(C341H481O40N5S6),研究了单个或多个干酪根分子模型在直接热解、加氢热解、蒸汽热解和氧气燃烧中的热解过程、产物分布及形成机理。而Zhao et al.[47]则是研究了过热蒸汽对于抚顺油页岩干酪根(C228H350O11N6S3)大规模体系的原位热解产物的影响。分子模拟是基于统计平均的,单个干酪根分子热解所产生的误差可能比较大[58],而由多个干酪根模型组成的大规模干酪根体系能够保模拟结果的准确性,这一点已经得到了证实[45]。
-
分子模拟方法主要分为两类,一类是包括从头算方法、半经验方法和密度泛函理论(Density Functional Theory,DFT)的量子力学模拟,这类方法可以精确描述分子之间的相互作用,但是仅适用于模拟几十到几百个原子的单一分子体系,速度较慢[1]。另外一种是包括分子力学(Molecular Mechanics,MM)、分子动力学(Molecular Dynamics,MD)、蒙特卡罗(Monte Carlo,MC)模拟和布朗运动(Brownian Dynamics,BD)的分子力学模拟,其以经典牛顿力学为基础[1]。这类方法在一定范围内接近量子模拟结果,适用于多分子系统,可达到上万个原子,并且速度较第一类方法更快,因此更适合研究像干酪根这种大分子体系的模拟。
随着计算机科学技术的发展,计算能力的提高使分子动力学模拟能够在较长的时间尺度上分析大规模体系的物理化学过程,这使得分子动力学模拟在干酪根的三维分子结构构建、热成熟演化、热解及其与矿物和气体的相互作用等相关研究中得到广泛的应用[32]。在分子动力学模拟过程中,最重要的部分之一是力场,它是用于计算模拟体系势能的计算方法。在干酪根模拟中常用的力场有Amber、Dreiding、PCFF、Compass和ReaxFF。其中,van Duin et al.[61]开发的ReaxFF力场是基于键级的反应力场,可以用来模拟反应体系中原子间键的断裂和形成过程,从而区别于其他的非反应力场。目前,研究者已经成功地将经典分子动力学与ReaxFF力场相结合形成反应分子动力学(Reactive Molecular Dynamics,RMD)模拟,来描述复杂体系中的化学反应变化。该方法已经证实了模拟煤热解的可行性和准确性[62⁃63],并且最近几年被扩展用于研究干酪根,以提高对热解机理的理解[14,55,59,64]。
由于计算能力的限制,热解化学反应的模拟时间必须远小于实验的实际时间。而根据阿伦尼乌斯公式,对于简单的反应,在一定的温度范围内影响反应速率的是温度,而不是活化能。因此,许多研究人员使用远高于实验的温度来加快模拟的速度,从而达到在短时间内研究模拟体系[14,64⁃65]。Salmon et al.[65⁃66]采用这种高温短时的方法对海藻胶生物聚合物分子和莫维尔褐煤分别进行了RMD模拟,发现海藻胶结构的热解机制开始于类异戊二烯侧链的断裂,之后酯官能团的C-O键断裂产生大量CO2。在较高的温度下,C2H4的生成是连续的β裂解反应的结果。生成产物的顺序是类异戊二烯结构、CO2和高温下的C2H4,这与实验结果[67⁃68]非常一致。同时,海藻胶结构的C40+模拟产物与实验残余物在H/C和O/C图版中的变化具有较强的一致性[65]。莫维尔褐煤的RMD模拟结果表明CO2是由羧基去官能团化产生的,这与NMR实验分析结果一致[66]。Hatcher et al.[69]基于对实际样品的热解实验提出了甲氧基中的C-O断裂产生甲基自由基,而甲基自由基可以参与多种反应,形成固态、液态和气态烃产物,而莫维尔褐煤的含甲氧基官能团模型的模拟结果验证了这种甲基形成机制[66]。产物生成的顺序是CO2、芳香族化合物和CH4,与褐煤的实验结果[70]一致。虽然两种大分子模型的RMD模拟和实际实验在温度和时间上存在很大差异[65⁃67,70],但RMD模拟对热解的产物及反应机制进行了再现和验证。由此说明,这种短时高温的模拟可以用于分析干酪根的热解过程以及形成机制。
针对干酪根热解生烃的分子模拟,其具体流程如下:(1)通过Materials Studio软件中的Amorphous Cell模块构建干酪根超晶胞模型;(2)对超晶胞模型进行几何优化、模拟退火、NPT系综动力学模拟使体系密度达到平衡、合理,获取各体系的晶胞参数,为后续热解模拟奠定基础;(3)根据干酪根热解所要研究的影响因素(温度、升温速率、压力等),在编写的In文件中设置参数范围。例如:Han et al.[71]对干酪根进行了等温热解模拟,其中设置了10个温度点(1 200 K、1 400 K、1 500 K、1 600 K、1 800 K、2 000 K、2 200 K、2 400 K、2 500 K和2 600 K),每个温度点模拟时间为500 ps;Qian et al.[14]对干酪根进行了300~3 300 K内的升温模拟,其升温速率分别设置为10 K/ps、20 K/ps、40 K/ps、60 K/ps和100 K/ps;Chen et al.[72]对正十六烷进行了等压模拟,其压力点设置为10 MPa、30 MPa、50 MPa、70 MPa和90 MPa;(4)通过Lammps软件进行RMD模拟;(5)热解后处理分析:使用Fortran语言编写脚本进行产物碎片统计分析、反应路径和轨迹文件分析。因此,使用分子模拟方法来探索干酪根热解生烃机理可以很容易实现超高温和超高压的模拟,并且安全、无污染、成本低、速度快,更关键在于可以实现对干酪根热解的瞬态中间产物或过渡态进行研究,为从原子分子层次认识干酪根有机大分子体系热解的反应机理提供了新的途径,这正是分子模拟相对于传统的生烃热模拟实验突出的优点之一。
-
在干酪根热解生烃过程中,温度、升温速率、水、压力、无机矿物质等因素对油气的生成具有重要的作用。因此,结合实验阐述了分子模拟研究下各个因素对于干酪根热解产物的影响及生烃机理。
-
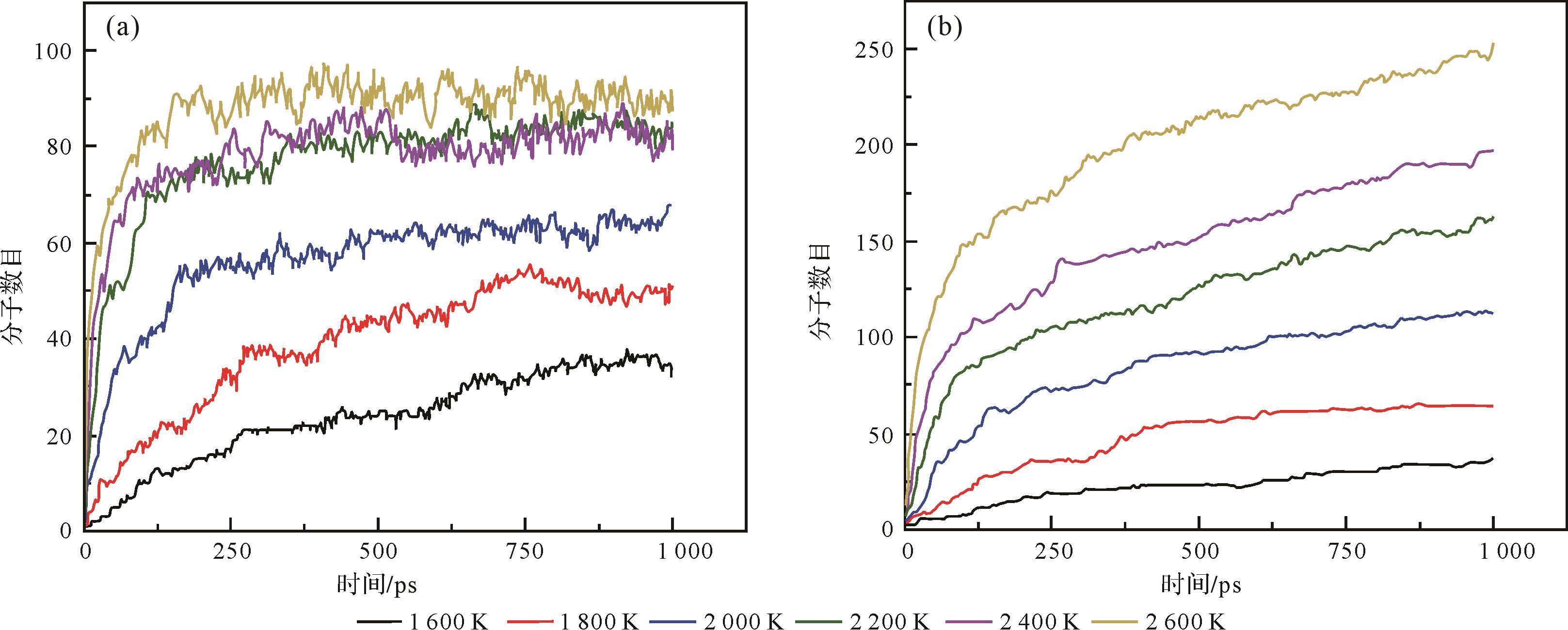
一般来说,干酪根热解的分子数随着温度的升高而增加[45,58⁃59],而高温下的分子数变化曲线相对于低温下出现了较大波动,表明高温下发生了部分的聚合反应[58]。此外,高温不利于直接热解(图3a)过程中反应的充分进行,而有助于加氢热解(图3b),这是因为在2 200 K、2 400 K和2 600 K的温度下,直接热解的分子数目达到了几乎相同的最大分子数目,而在加氢热解过程中随着温度的升高,分子数目不断增加[59]。

Figure 3. Time evolution of molecules produced by pyrolysis in the Longkou oil shale kerogen at six different temperatures during direct pyrolysis (a) and hydropyrolysis (b) (modified from reference [59])
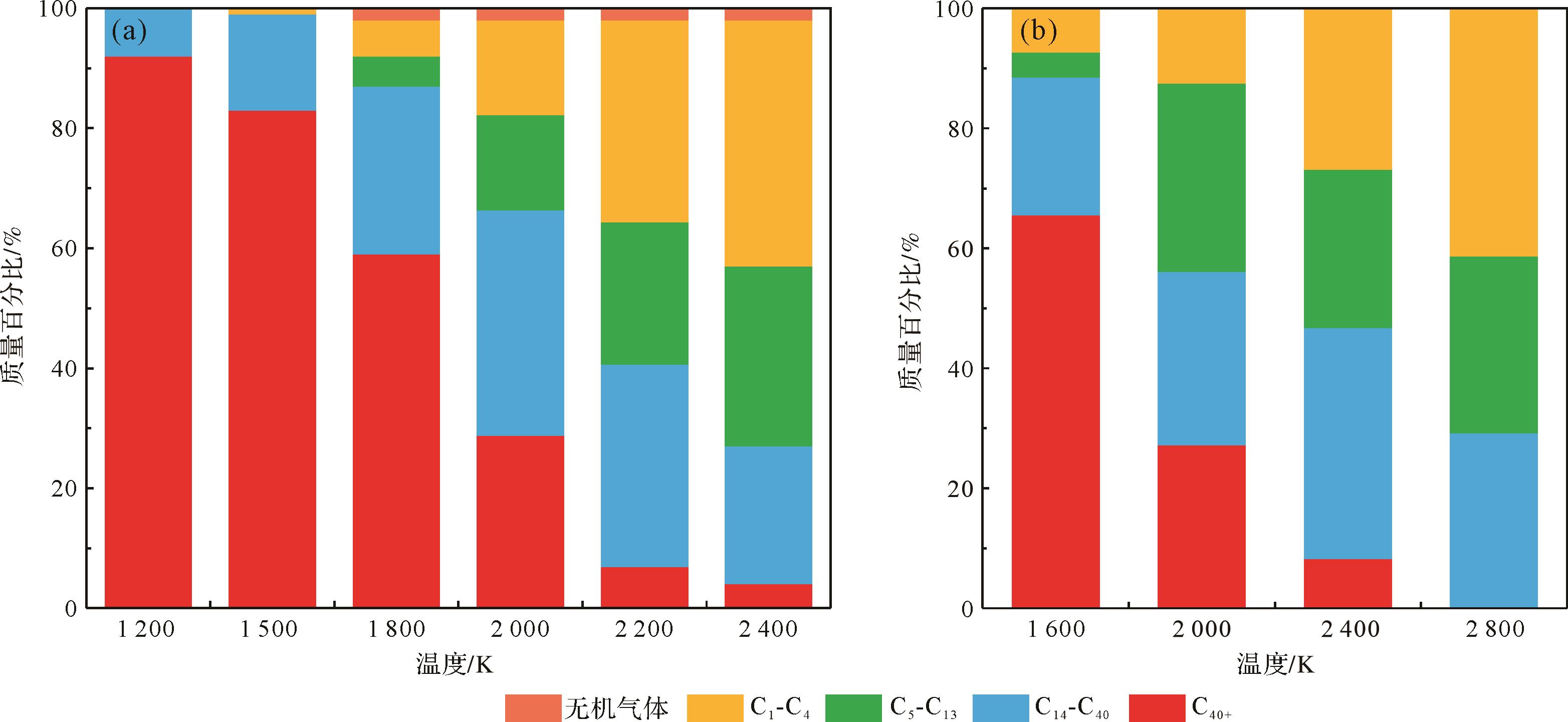
根据碳原子的数量,油页岩干酪根热解产物可分为四类[64]:C1-C4、C5-C13和C14-C40分别被认为是天然气、轻质页岩油和重质页岩油,而C40+被视为焦碳。Liu et al.[64]和Zhang et al.[58]分别对绿河油页岩I型干酪根模型和龙口油页岩II型干酪根模型进行最高温度为2 400 K和2 800 K的等温热解模拟,如图4所示,随着温度的升高,C1-C4气体的含量逐渐增加,而C40+化合物则逐渐分解成小分子,在高温下没有或只有少量的C40+化合物存在,这与实验上发现气态碳氢化合物(C1-C4气体)在低温和高温下的比例均增加[73],并与干酪根在黄金管限定体系中发生初次反应的变化趋势一致[74⁃76]。与上述不同的是,重质页岩油(C14-C40)随着温度的升高首先分别在2 000 K和2 400 K下达到峰值,然后再逐渐降低。这种利用RMD方法得到的重质页岩油产率随温度的演变趋势与实验得到的结果一致[3,74,76⁃77]。Cao et al.[76]发现轻质页岩油(C5-C13)的变化趋势也是如此。徐金泽等[78]认为这是由于随着温度的升高,干酪根的初次热解逐渐完成,但是过高的热解温度会导致页岩油发生二次裂解生成气体,降低页岩油产率。在龙口油页岩II型干酪根加氢热解和直接热解过程中,分别在2 000 K和2 200 K获得最佳的轻质页岩油产率[59]。根据Han et al.[71]建立的干酪根热解分子模拟的成熟度标尺可知,在地质条件下龙口油页岩II型干酪根在Ro为0.96%和1.25%时达到最大的轻质页岩油产率,为获取地质条件下的最大轻质页岩油产率提供了途径。因此,通过分子模拟手段研究干酪根热解生烃对于地质条件下的生油产率具有一定的指示意义。

原位转化工艺(ICP)是提高页岩储层油气采收率的一种方法,其目的是通过产生发育良好的孔隙网络来促进地下页岩干酪根的热解速率并提高页岩基质的渗透性。目前,ICP技术的一个关键挑战是最佳加热温度的选择。Xu et al.[56]采用RMD模拟方法研究了不同成熟度的干酪根(II-A、II-B、II-C、II-D)[39]在不同加热温度(1 500~3 000 K)下的热解行为,发现干酪根热解孔隙网络的结构特征取决于干酪根的成熟度和热解温度。通过比较不同温度(1 500 K、2 000 K、2 300 K和3 000 K)下不同成熟度干酪根模型的孔隙网络特征,发现升高温度均有利于不同成熟度干酪根形成更好的热解孔隙网络[56]。然而,不同成熟度的干酪根模型对孔隙网络特征表现出不同的温度响应。低成熟度干酪根(II-A、II-B)的热解孔隙—网络优于中、高成熟度干酪根(II-C、II-D)。对于低成熟度干酪根(II-A、II-B)而言,热解模拟的最佳热解温度约为2 300 K,此时干酪根孔隙网络的孔隙率、孔径、比表面积等参数值达到最大[56]。通过阿伦尼乌斯公式建立的RMD模拟温度与ICP工程温度之间的转换关系(公式1)[56],可以得到ICP工程的最佳加热温度为730 K左右,进而从分子模拟角度为ICP工程的最佳温度提供了建议。未来针对特定的页岩储层,需要考虑更多因素来确定ICP的最佳加热工程温度,包括干酪根类型、成熟度阶段、热导率、加热时间等[56]。
(1) 式中:Teng为ICP工程温度(K);Tsim为模拟温度(K);ICP工程的加热时间teng=103 s;模拟时间tsim=10-9 s;干酪根活化能Ea值介于55~65 kcal/mol;kB为玻耳兹曼常数。
值得注意的是,高温RMD模拟的时间尺度远远短于干酪根在深成热解作用期间发生缓慢热化学变化所对应的地质时间尺度(几到十几个百万年),这将不可避免地对模拟结果产生一些影响。一方面,会大量生成C2H4而造成产物组成差异性较大[79];另一方面,由于模拟时间比实际热解时间短很多,导致了干酪根热解产物中产生大量中间产物,而导致产物类型复杂多变[64,80]。而与传统的MD相比,Wang et al.[44]采用混合分子动力学/力偏蒙特卡罗(MD/fbMC)方法在实验温度下(800 ℃和1 100 ℃)对松辽和鄂尔多斯干酪根进行了0.1 ns的热解模拟,模拟结果表明甲烷、其他气体产物和残渣的质量分数与实验结果一致。Atmani et al.[35⁃36]报道了副本交换分子动力学(REMD)的方法可以在接近地质条件(150 ℃和10~100 MPa)下进行RMD模拟。这为将来在实验温度和地质条件下进行干酪根热解提供了可靠的模拟方法。
-
目前干酪根热解模拟的研究方法大多数都是等温法,然而,与等温模拟相比,非等温模拟有许多优点:(1)在足够低的升温速率下的升温模拟可以连续观察干酪根热解过程中产物随温度和时间的变化,因此更接近于模拟地质条件下油页岩干酪根的实际热解反应过程[81⁃83]。(2)非等温模拟在计算上更省时。如Zhang et al.[84]通过GMD-Reax计算比较了木质素的等温和非等温热解模拟,发现模拟时间为4 500 ps的缓慢升温模拟(0.4 K/ps)需要714 h,比类似温度范围的一系列等温模拟需要2 500多个小时要快得多。(3)非等温热解模拟仅需模拟一个热解过程就可以在较大温度范围内研究温度的影响[82]。
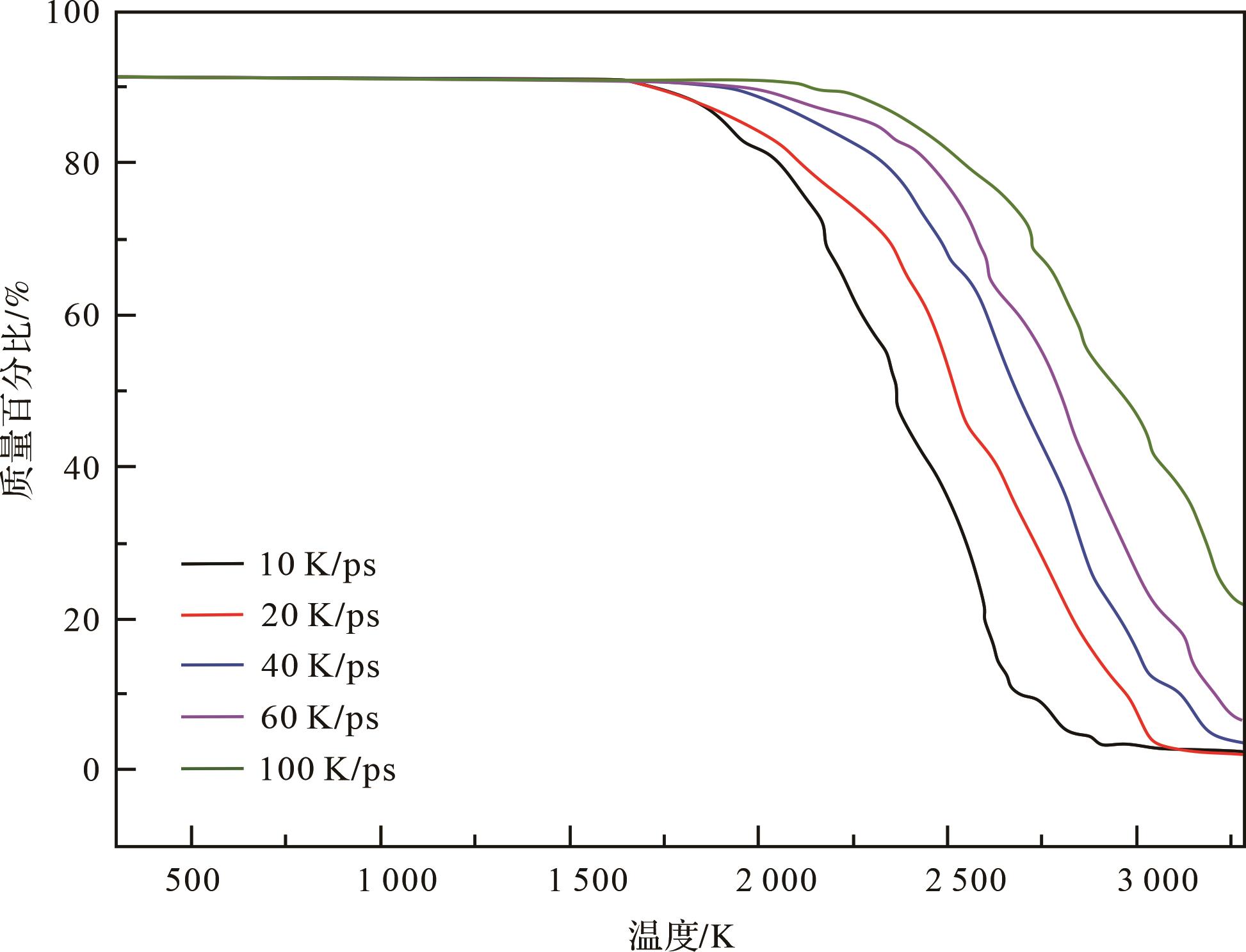
Qian et al.[14]对绿河油页岩I型干酪根模型进行了非等温热解模拟。如图5所示,可以看出,升温速率的增加会使干酪根开始热解的温度升高,这与干酪根的热重分析(TG)实验[4]和基于Ungerer et al.[39]建立的II-C型干酪根模型的热解结果一致[81],并且在煤的热解中也得到了证实[82]。

Figure 5. Weight loss curves of C40+ compounds during the pyrolysis of Green River oil shale kerogen at five different heating rates (modified from reference [14])
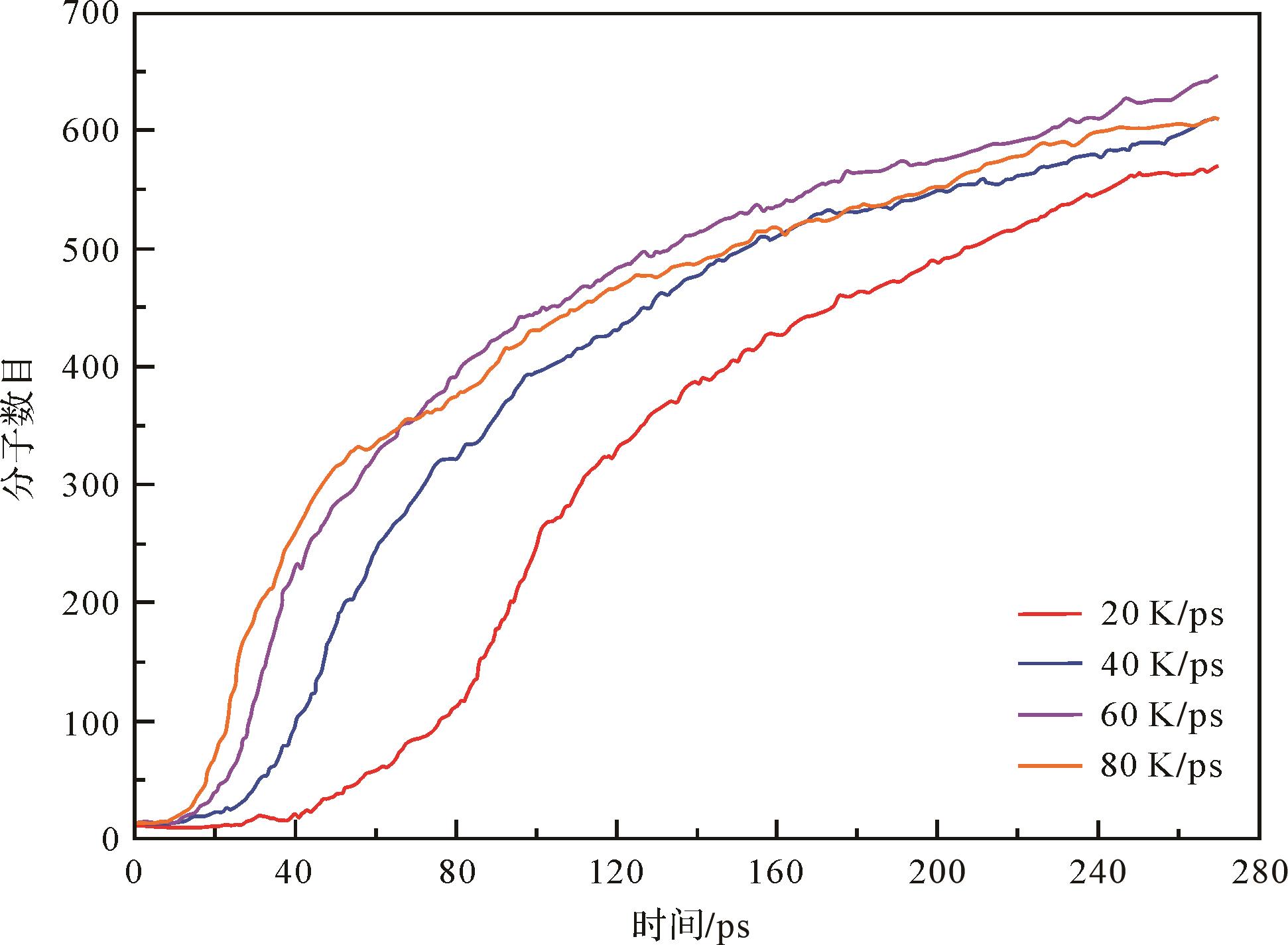
Zhang et al.[85]模拟了桦甸油页岩干酪根模型在20 K/ps、40 K/ps、60 K/ps、80 K/ps四种升温速率下的加氢热解过程,如图6,干酪根热解产生的分子数目随着升温速率的增加而增加,但是过高的升温速率(80 K/ps)会降低热解产生的分子数目,并且其产物焦炭(C40+)只能在80 K/ps的加热速率下观察到,这是因为升温速率越高,反应时间越短,这可能导致干酪根在很高的升温速率下不完全热解。在升温速率为60 K/ps以下,页岩油产物(C5-C13和C14-C39)随着升温速率的增加呈现平缓增加的趋势,而在80 K/ps时有所降低。由于油页岩工业利用过程中的主要产物是页岩油,因此桦甸油页岩干酪根加氢热解的升温速率应控制在60 K/ps以便生成更多的页岩油产物[85]。分子模拟上得到的页岩油产率与升温速率之间的变化趋势与实验上油页岩热解得到的认识是相似的。Han et al.[86]研究表明升温速率低于10 ℃/min时,升温速率与页岩油产率之间呈正相关关系。然而,升温速率高于10 ℃/min的热解可能是由于油页岩气孔在较高升温速率下发育时间不够,产物生成速度大于从所处孔隙中逸出的速度,导致页岩油发生二次焦化反应,产率降低,生成焦炭[87⁃88]。Wang et al.[5]在实验上研究了升温速率(5~20 ℃/min)对于桦甸油页岩热解产物的影响。结果表明随着升温速率的增加,页岩油产率先增加后略有下降,在升温速率为12 ℃/min时,页岩油达到最高为15.696%的产率。

Figure 6. Time evolution of molecules during pyrolysis of the Huadian oil shale kerogen at four different heating rates (modified from reference [85])
干酪根的等温热解和非等温热解分子模拟表明,在相对较低的温度下,页岩油产率随着温度的升高而增加,但当温度很高导致页岩油发生二次反应时,页岩油产率反而会降低。同时,较高的页岩油产率也可以通过较高的升温速率获得,除非该速率导致极短的反应时间,从而导致干酪根的不完全热解,使得页岩油产率降低。这与Qian et al.[63]对次烟煤进行等温热解(1 600 K、2 000 K、2 400 K和2 800 K)和非等温热解(20 K/ps、40 K/ps、80 K/ps和100 K/ps)模拟得到的结果一致,并且他们还发现焦油质量随温度升高而变好,但是随着升温速率的提高而变差。因此,仅通过温度或加热速率不足以明确页岩油/焦油预期的产率高峰。
-
Lewan et al.[89⁃91]通过有水与无水参与的热模拟实验对比研究表明,有水参与的反应体系产物特征相对无水参与的反应体系能更好地与实际地质体中烃源岩的热演化产物进行对比。近两年,少部分研究者研究了干酪根在水分子的参与下的模拟热解过程以及产物。Zhang et al.[46]对龙口油页岩干酪根进行了直接热解和水蒸气热解的RMD模拟,发现蒸汽热解下H2O分子可以提供更多的氢自由基参与反应,从而促进了干酪根和重质页岩油的裂解,阻碍了C-C交联结构的形成,提高了轻质页岩油(C5-C13)和气体(C1-C4)的产率。这与实验上水对油页岩热解[88⁃89,92]和Zhao et al.[47]研究过热蒸汽对于抚顺油页岩干酪根原位热解产物的模拟结果一致,并且他们还得出H2O有助于提高页岩气(C1-C4)和轻质页岩油(C5-C13)的饱和度,从而提高热解产物的质量。
对于干酪根蒸汽热解气体产物而言,H原子的含量略有增加,而O原子的含量几乎是直接热解的两倍[46],这与使用RMD模拟研究褐煤[93]和木质纤维素生物质[94]的蒸汽热解结果类似。相比之下,蒸汽热解产物页岩油中的H原子含量与直接热解相同,而O含量在蒸汽热解后略有下降,杂原子N和S在轻质页岩油和气体中的占比增加。这些结果表明,蒸汽热解可以促进氧和杂原子形成轻质产物[46]。
需要注意的是,目前分子模拟上研究水对干酪根热解的影响时,是采取向干酪根分子中加入一定数量水分子的方法,并没有考虑水相态的转变以及可能包含无机盐、金属离子等的实际地层水对于干酪根热解的影响。这是未来从分子模拟角度研究水对干酪根热解影响需要考虑的方面。
-
压力对有机质生烃、热演化的影响一直备受争议[95⁃96],促进作用、抑制作用和无明显作用是最常见的三种认识,其中抑制作用被广大学者所接受。例如,郝芳等[96]通过实际盆地资料研究表明超压对有机质演化和生烃具有抑制作用。此外,油页岩热解的高压实验分析表明,随着热解压力的增加,抑制了油的蒸发,促进了二次裂解反应,从而使得石油产量降低,天然气产量增加[2,97⁃98]。Chen et al.[72]基于RMD研究了不同压力对于正十六烷裂解的影响,模拟结果表明产物H2分子的数量和压力之间呈正相关,而增加压力对乙烯的形成有阻碍作用,这与先前的实验研究一致[99]。目前,通过分子模拟手段研究压力对于干酪根热解生烃产物以及机理的成果比较少,未来关于压力对干酪根热解分子模拟的研究是一个很好的发展趋势。
在实际地质条件下,沉积岩中的干酪根是不连续地分布在无机矿物质当中,诸如黏土矿物、石英、白云岩、方解石、伊利石、黄铁矿等矿物以及一些微量元素Fe、Cu、Ni、Zn等,这些无机矿物质对干酪根热解生烃具有重要的影响。茹鑫[19]和Hu et al.[11]对桦甸油页岩干酪根进行无水热解实验,发现蒙脱石和石膏对于干酪根热解具有催化作用,能增加产油率,降低结焦率;高岭石是弱催化剂,而方解石则是抑制剂,会阻碍干酪根的生油过程;蒙脱石、高岭石的存在使得干酪根热解产物中烯烃减少,饱和烃增加,热解烃中先前生成的C12+部分转化为低分子烃类(C7-C12),使低分子烃类富集。Ma et al.[13]发现在蒙脱石、黄铁矿、铁及其混合物对III型干酪根具有明显的催化作用,促进了烃类气体生成,其催化效率分别为:干酪根+混合催化剂>干酪根+黄铁矿>干酪根+蒙脱石>干酪根+铁>干酪根。然而,利用分子模拟技术研究无机矿物质对干酪根热解影响的成果相对较少,原因是在矿物存在情况下研究干酪根热解时缺乏适用于矿物的反应力场参数,而这正是未来的干酪根热解分子模拟研究的一个重要方向。
-
干酪根热解会产生大量烷烃、烯烃和芳烃产物,其中烷烃和烯烃是页岩油和页岩气的主要成分[14,19,64]。由于短链气态烃(C1-C4)、H2、CO2、CO、H2O等是页岩气的重要组成部分。因此,产物类型及其分子数量变化是目前研究热点。
Zhang et al.[85]对桦甸油页岩干酪根进行了加氢热解分子模拟,结果表明高升温速率明显促进了CH4和H2O的生成,在80 K/ps的升温速率下达到最高产率[85]。然而,Nazzal[88]对约旦油页岩进行热解实验发现,不同升温速率(2~30 ℃/min)对于氮气/蒸汽环境中H2O的产率影响并不大,这与分子模拟得到的认识有所差别。
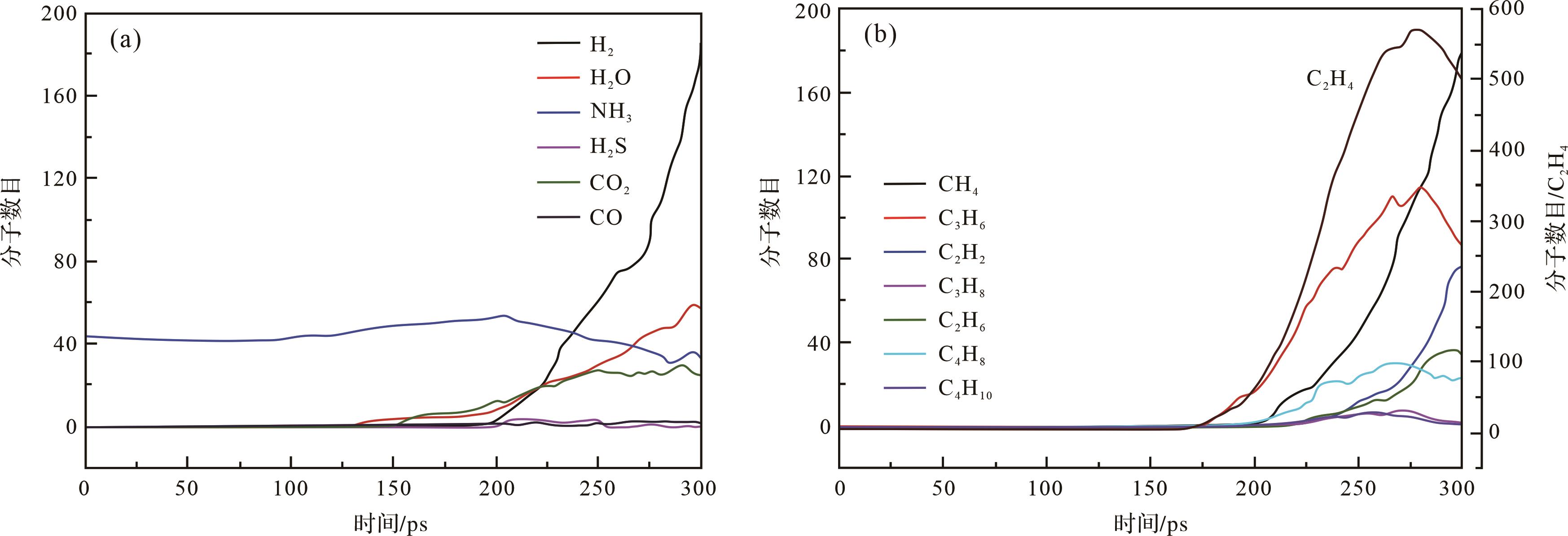
以10 K/ps升温速率对绿河油页岩I型干酪根进行分子热模拟,结果表明无机气体中CO2和H2O优先生成,并随着温度的升高而增加[14](图7a),这与Fletcher et al.[100]的实验结果一致;约200 ps之后H2产率迅速增加,而CO、H2S的分子数量则一直处于一个较低水平。在生成有机烃的过程中,优先生成C2H4、C3H6;其次是CH4,其产率随着温度的升高而快速增加(图7b)。而C2H4、C3H6、C2H6、C4H8等典型有机气体在后期热解过程中发生了二次裂解,导致H2、CH4以及C2H2气体的产率增加[14],这与褐煤[82]和次烟煤[63]的非等温热解结果一致。

Figure 7. Time evolution of molecules of inorganic (a) and organic gas (b) produced by kerogen pyrolysis at the heating rate of 10 K/ps (modified from reference [14])
比较有机和无机气体生成顺序可以发现,CO2和H2O是最先产生的气体,其次是C2H4、C3H6和CH4,这与次烟煤在分子模拟(20 K/ps)和TG-MS(5 K/min)实验分析下得到的结果一致[63]。此外,RMD模拟得到的无机气体产率H2O>CO2>CO、有机气体CH4和C2-C5的变化也与约旦油页岩[88]、绿河油页岩[101]和青山口组I型干酪根[76]、嫩江组II型干酪根[102]在实验上的热解趋势是一致的。总体来说,热解产物类型与绿河油页岩热解实验所得到的气体产物类型基本一致[100⁃101,103]。但是,无机气体H2和有机气体C2H4的数量远远高于其他无机有机气体,这与实验条件下[5,76,88]所得到的结果明显具有较大差别。由此说明,干酪根的热解是一个多因素的过程,不仅与温度和压力有关,还与矿物组分、外来氢源等因素有关。分子模拟作为一种重要的研究手段,从微观上对干酪根的裂解过程及其热解产物,如页岩油组分(C14-C40和C5-C13)、有机气体(CH4、C2H4)和无机气体(CO2、H2、H2O、H2S)的形成机理提供了一定的认识。
干酪根是由C-C、C=C、C-O、C-N、C-H、C-S等多种化学键不规则连接的有机大分子,其热裂解过程中键的断裂基本符合最小键解离能(BDE)原则[14,45,64]。Guan et al.[104]基于密度泛函理论(DFT)计算了554个干酪根模型片段中化学键的键解离能(BDE),由于化学环境的不同,每一种化学键的BDE值变化范围较大且相互重叠,但BDE值的基本变化趋势是C-S<C-N<C-O<C-C<C-H<C=C。然而,BDE除了取决于化学键的化学环境外,还取决于自由基的稳定性,这为干酪根热裂解过程中键断裂的顺序提供了参考。
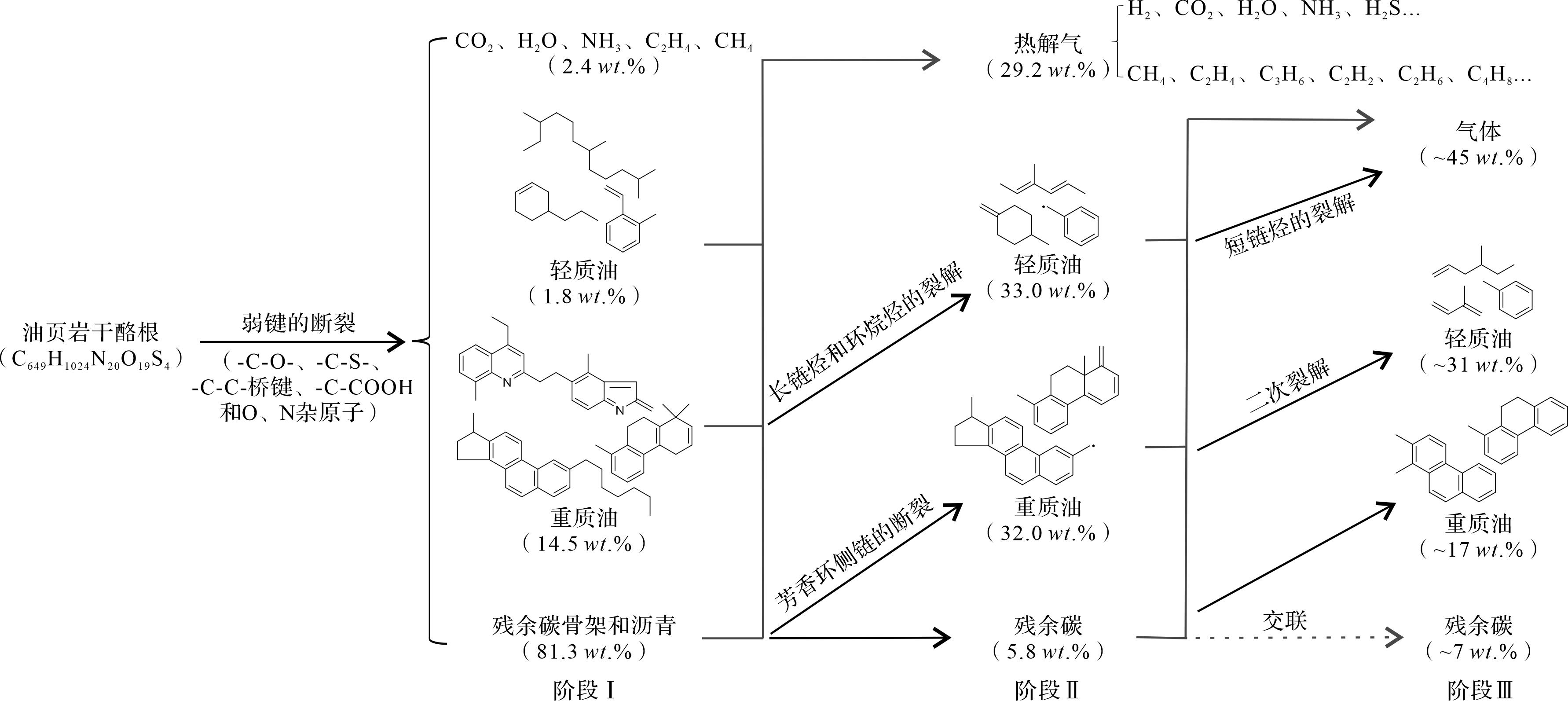
Qian et al.[14]和Liu et al.[64]基于绿河油页岩I型干酪根模型[37]分别进行了300~3 300 K的升温和1 200~2 400 K的等温RMD模拟,进而分析干酪根的热解机理。根据绿河油页岩干酪根在升温模拟过程中的C40+化合物的变化、键断裂特征、典型产物分布和反应路径,Qian et al.[14]将油页岩干酪根的整个热解过程分为三个阶段(图8)。阶段I,干酪根骨架结构中的-C-O-、-C-S-、-C-C-桥键、-COOH和O、N杂原子等弱键发生裂解,生成部分重质油、轻质油和少量的CO2、H2O、NH3、C2H4和CH4等气体;阶段II,芳香环侧链的断裂以及环烷烃和长链烷烃发生裂解生成大量的重质油和轻质油产物;阶段III,自由基的交联、重质油二次裂解和短链烷烃的进一步裂解导致焦炭生成、重质油减少和以H2为主的含氢气体急剧增加,对应于干酪根在高温或长时间热解实验的初始结焦阶段。干酪根在阶段I和II的初始热解机理与Liu et al.[64]的结论是一致的,并且他们还得出芳香环上的开环反应较难进行,而侧链容易断裂形成BDE较低的苄基;多环烷烃中C-C键的断裂会引发一系列反应,包括开环反应、脱甲基反应和电子重排,而开环反应优先发生在与双键的间位;链状脂肪族碳氢化合物中C-C键相继发生断裂,生成小分子。

Figure 8. Pyrolysis process of the Green River oil shale kerogen and molecular structure of typical products (modified from reference [14])
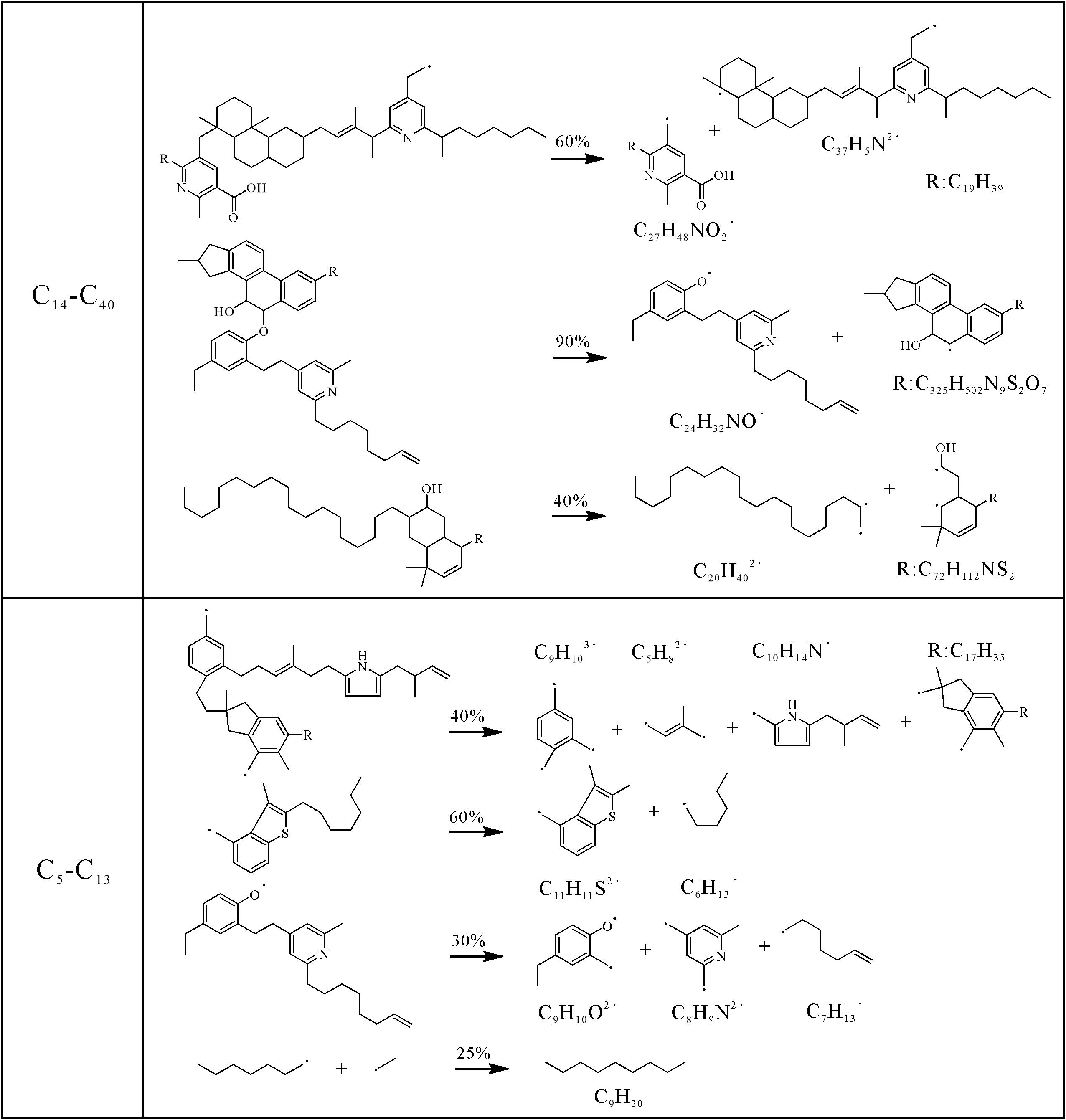
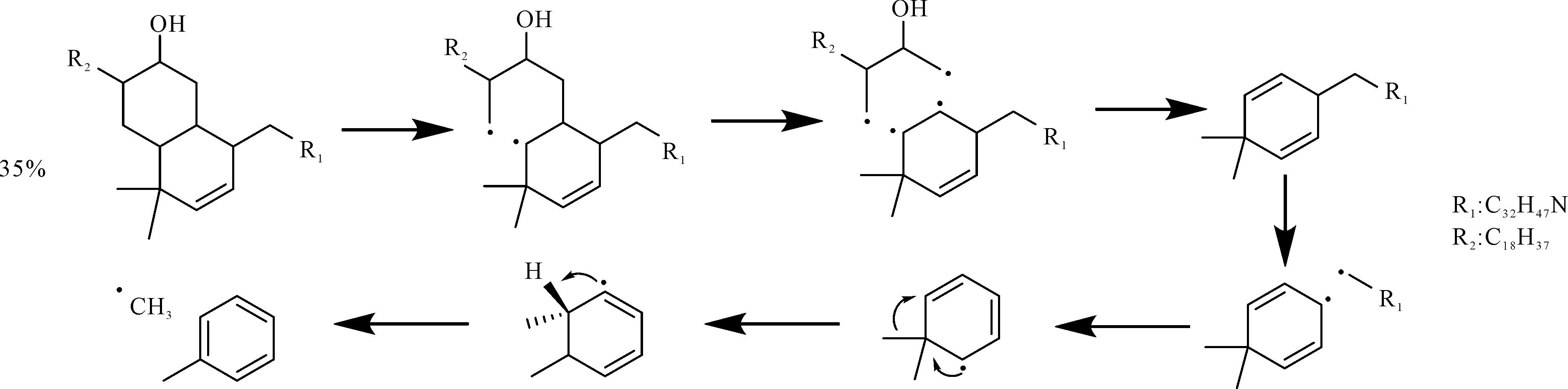
Liu et al.[64]阐明了绿河油页岩干酪根典型页岩油产物(C14-C40和C5-C13)的形成途径。如图9所示,生成C14-C40组分的典型分解反应包括C-C和C-O键的断裂,而C5-C13组分可以通过大片段的C-C键断裂或通过自由基之间的结合形成。十六烷作为柴油中最重要的成分,通过模拟观察到了形成十六烷的中间体-C16H33·自由基。另外,大量的芳香族结构存在于页岩油中。绿河油页岩干酪根RMD模拟过程中得到了典型单环芳香族化合物(甲苯、1-甲基-2-乙烯基苯、3-乙基-5-甲基苯酚)的形成途径。以甲苯为例(图10),其是通过双环结构先发生开环反应,之后自由基的重排并且官能团断裂而形成的。因此,通过分子模拟可以清晰地展现产物的中间体以及微观形成机制,更好地揭示干酪根的生烃微观机理。然而,由于页岩油产物类型和反应途径复杂,目前人们主要关注于页岩气的形成机制,而对于页岩油的生成机理研究还较少,缺乏系统的认识。

Figure 9. Reaction pathways of typical C14⁃C40 and C5⁃C13 components of Type I kerogen from the Green River oil shale at temperatures of 1 800 K, 2 000 K, 2 200 K and 2 400 K

Figure 10. Pathway of toluene formation from type I kerogen of the Green River oil shale in the Reactive Molecular Dynamics(RMD) process
CH4是干酪根热解的主要产物,其形成主要由甲基自由基和氢自由基的结合[45,58⁃59,64,85]。Qian et al.[63]对次烟煤进行RMD模拟发现其中甲基的形成有三种途径:(1)甲基醚(-O-CH3)断裂生成甲基(·CH3);(2)芳族侧链(-Ar-CH2-CH3)断裂成甲基;(3)链烃(-(CH2)n-CH3)断裂成甲基。长7段II型干酪根热解模拟产生的CH4中氢自由基来自于甲基[45],这与龙口油页岩II型干酪根模拟中氢自由基来自羟基不同[58⁃59],主要是因为在长7段II型干酪根体系中甲基比羟基更丰富。在RMD模拟中观察到的大量C2H4分子可以通过长链脂肪烃的C-C断裂生成乙烯,它也可以由C2H5·脱氢生成[14,63⁃64]。
对于无机气体来说,CO2的形成与脱羧基过程有关[14,58,64,66,85],这在干酪根热解实验中已经被证实[2,70]。在干酪根热解中,H2主要由烃类热解产生的氢自由基两两发生碰撞而形成[64]。Yang et al.[105]在研究过热蒸汽实验下油页岩干酪根的原位热解机理过程中发现H2的形成机制尚不明确,而Zhao et al.[47]通过对抚顺油页岩干酪根模型进行RMD模拟,深入揭示了过热蒸汽下H2的三种形成机制:(1)均来自干酪根的两个氢自由基形成H2;(2)分别来自过热蒸汽和干酪根的两个氢自由基形成H2;(3)均来自过热蒸汽的两个氢自由基形成H2。H2O分子的形成过程是通过羟基与氢自由基结合而成[14,64],而其中氧和氢自由基的来源因干酪根模型和热解模拟方式的不同而有较大差异。例如,龙口油页岩II型干酪根在直接热解生成的第一个H2O分子来源于醚基的氧和N-H键的氢,而在加氢热解过程中H2O分子中的氧和氢分别来源于羧基和H2[59];长7段油页岩II型干酪根直接热解生成的H2O中的氢自由基来自于C-H键,而不是N-H键[45]。
硫原子在热解过程中转移到气体产物中是液体产物脱硫的过程,其产生的气体产物主要是H2S。在龙口油页岩II型干酪根直接热解过程中,噻吩和亚砜均是由先形成的巯基(HS·)与H2O分子反应生成H2S分子,而在加氢热解过程中,两个化合物在H2分子的作用下先形成硫自由基,之后分别与H2和H2O分子发生反应形成H2S分子[59],这与长7段油页岩II型干酪根是通过六元环支链上的巯基自由基(HS·)捕获另一个六元环上羟基(HO·)中的氢自由基从而形成H2S分子的机理是不同的[45]。
4.1. 温度对干酪根热解的影响
4.2. 升温速率对干酪根热解的影响
4.3. 水对干酪根热解的影响
4.4. 其他因素对干酪根热解的影响
4.5. 干酪根热解的生烃机理
-
(1) 采用实验分析方法所构建的干酪根模型并不能很好地反映其孔隙结构,并且大部分干酪根模型分子量偏小,不具有足够的代表性。未来在构建方法上可以尝试机器学习法来构建大尺度的干酪根分子模型,相对分子量可达上百万;同时,所构建干酪根模型的元素含量、官能团组成以及微孔隙等尽可能地反映实际地质条件下的干酪根特征。
(2) 在实际地质干酪根模型构建方面,大部分均考虑纯有机质的干酪根模型,未考虑页岩矿物组分、地层水、有机酸以及无机盐等因素的影响。因此,分子模拟热解得到的规律与实验结果有所差别,对地质概况难以定性描述。未来,建立富含页岩矿物组分、地层水、有机酸以及无机盐的多尺度复杂干酪根模型将更加真实地反映实际地质概况。
(3) 通常采用提高模拟温度的方法来弥补地质上的热演化时间,进而缩短模拟反应时间,这是目前分子模拟的缺点之一。此外,干酪根的高温模拟会产生大量的C2H4,这与实验事实和地质概况不符。因此,未来发展低温热模拟方法来预测干酪根的热解规律是弥补实验—地质—理论的关键技术之一。值得一提的是,升温速率、水的相态、地层水、压力、矿物组分以及不同热演化程度对干酪根热解的认识也不充分,需要更为深入的研究来揭示它们对干酪根热解的影响。
(4) 目前,大多数研究工作主要致力于干酪根高温热解的气体产物,而对低温页岩油的生成机理缺乏系统的认识,这归因于低温热解反应较慢、产物类型复杂多变。因此,低温生烃机理的研究仍存在巨大的挑战,明确其热解机理对于页岩油的勘探与开发具有重要的指导意义。

 DownLoad:
DownLoad: